RNAhub
Background on non-coding RNAs and covariation analysis
2. GLY1 3′ UTR using the flanking mode
3. HOTAIR domain 1: long non-coding RNA without conserved structure
4. CWC15: example of protein-coding mRNA sequence
Motivation
Accurate RNA multiple sequence alignments are crucial for understanding RNA structure and evolution, as well as advancing next-generation deep learning methods for RNA structure prediction, such as those inspired by AlphaFold. However, generating these alignments and evaluating their quality remains a challenge.
We introduce a web server that is an online implementation of the pipeline introduced before by Gao et al. 2021 and focuses on rigorous homology search, providing accurate alignments with predicted secondary structure subjected to comprehensive quality assessments.
This web server has been designed to be user-friendly, allowing those without bioinformatics expertise to query a sequence of interest to determine if there is support for conserved RNA secondary structure.
Background on non-coding RNAs and covariation analysis
Many non-coding RNAs function through a conserved structure that allows for interactions between other RNAs, proteins, and metabolites. For example, transfer RNAs (tRNAs), which are critical for translation, adopt a cloverleaf secondary structure (Watson Crick or wobble base pairing) and L-shaped tertiary structure. This structure allows the tRNA to engage with the protein subunits of the ribosome in a protein-RNA complex. As structure is critical to the function of structural non-coding RNAs, these non-coding RNAs display a distinct evolutionary signature that favors the maintenance of structure via compensatory base pair substitutions. Compensatory base pair substitutions refer to two nucleotide substitutions across a distance of evolutionary time that maintain RNA secondary structure even as the primary sequence changes. For example, in one species, a base pair could be A-U while in another species, it would be G-C; in either case, the base pairing interaction is maintained. These compensatory base pair substitutions provide evidence in favor of an evolutionary constraint to maintain RNA secondary structure.
As compensatory substitutions occur across millions of years of evolution, multiple sequence alignments of homologous sequences have to be built across species. Once an alignment is built, the R-scape method (Rivas et al. 2017) identifies pairs of covarying positions in the alignment that significantly covary with each other. R-scape’s null model accounts for covariation that can result from the phylogenetic relationship of the sequences in the input alignment.
In Rivas et al. 2017, we demonstrated that R-scape identifies significant covariations in many known structural RNAs while failing to detect them in some long non-coding RNAs such as HOTAIR. When the tunable expectation value (E-value) is set to a stringent value such as the default of 0.05, R-scape is very specific and less prone to false positive base pairs than other methods. In Gao et al. 2021, we demonstrated that R-scape can be used to identify evidence of conserved RNA structure for a query sequence for which we do not know whether a structural RNA is present. In RNAhub, we developed a workflow in which a query sequence is searched iteratively against a database of choice for homology, resulting in a final alignment that is passed to R-scape to identify significantly covarying base pairs. These base pairs, which have many compensatory base pair substitutions, provide evidence in favor of an RNA whose function is dependent on its structure.
This RNAhub web server builds upon the structural homology searcher from Gao et al. 2021 in the following ways:
- Easy-to-use web server for performing all computations
- Ready-to-go genome databases to homology search the query sequence against
- Additional documentation and user flexibility
Method overview
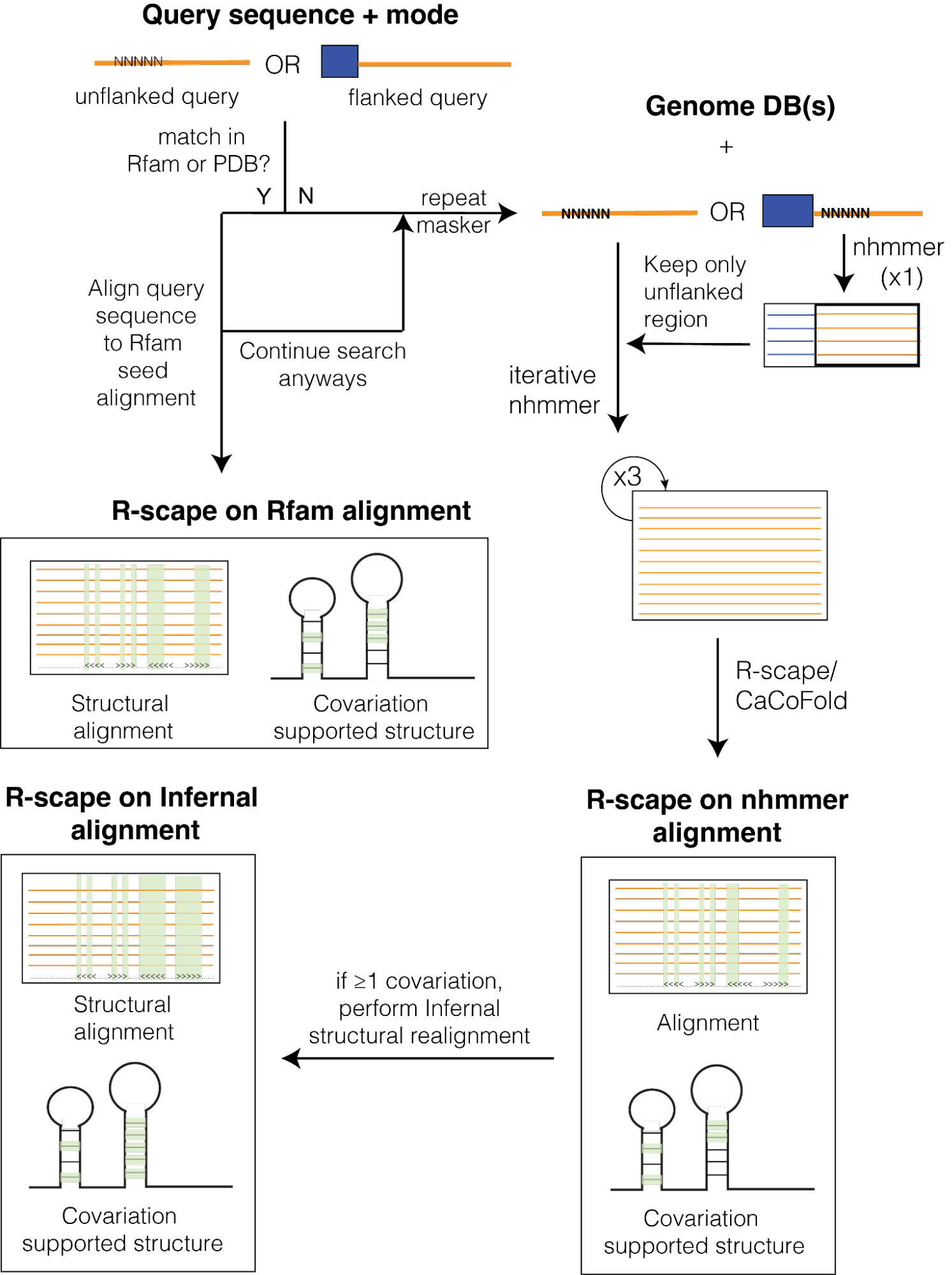
The query sequence must be a single DNA/RNA sequence. First, the server searches the Rfam database. If there is a hit to the Rfam database, the query sequence and seed sequences from Rfam are used to create a final alignment. In this alignment, the query sequence is integrated into an alignment with the Rfam seed sequences, and according to the Rfam annotated structure (using a covariance model). R-scape is then run on this alignment and a covariation-optimized structure is produced.
If no hit is detected to Rfam, three iterative homology searches are performed using nhmmer. The final alignment obtained after three iterative homology searches is evaluated using R-scape, a method that looks for evidence of conserved RNA structure by measuring pairwise covariations (compensatory base pair substitutions) observed in the alignment. The server provides R-scape results listing base pairs with covariation support and an R-scape/CaCoFold secondary structure, which integrates all covariation signals to improve structure prediction.
There are two modes implemented in RNAhub: unflanked and flanking. The unflanked mode requires a single query sequence and will produce an alignment based on the query sequence and then run R-scape on the final alignment.
In contrast, the flanking mode first performs an alignment using a flanking region to the query sequence, which the user must specify. After performing one iteration using the flanking region, the rest of the mode is the same as the unflanked mode. The flanking mode is often advantageous in cases where the query sequence is located within an intron or untranslated region (UTR) of an mRNA. Since the primary sequence of translated coding sequences is typically more conserved at the primary sequence level than introns and UTRs, we recommend using a neighboring exon as a flank if the query sequence is an intron or UTR. We illustrate an example using the flanking mode on the 3′ UTR of GLY1 that uses the adjacent single coding sequence of GLY1 as the flank.
Databases
We provide several ready-to-go genome databases to search the query sequence against. These genome databases were downloaded using the NCBI command line interface for downloading genomes.
- Metagenomes: metagenomic database of cultivated and uncultivated viral sequences (see Camargo et al. 2023)
- Viral: All available NCBI viral sequences. This is a much smaller database of better-characterized sequences than the metagenomic database.
- Bacteria: All NCBI reference + annotated Bacteria genomes.
- Fungi: Subclustered NCBI reference fungal genomes
- Metazoa: Subclustered NCBI reference invertebrate and vertebrate genomes
- Plants: Subclustered NCBI reference plant genomes
Eukaryotic genome databases (Fungi, Metazoa, Plants) were clustered into groups based on the primary sequence of their RNase MRP sequences, and one genome per cluster was kept to create a smaller subclustered genome database. This was done to significantly reduce the time required to decrease the genome database size for producing alignments while also maintaining enough genomes to detect significant covariations.
Examples of how to use RNAhub
Four distinct examples and modes of RNAhub are demonstrated.
The first example is to use a full search using the unflanked mode to obtain an alignment and predict secondary structure (including the correct prediction of the pseudoknot). In this case, there is a hit to Rfam. The user can either continue the standard RNAhub approach or use the Rfam seed alignment to directly align the query sequence.
The second example uses the flanking mode to improve homology search, allowing for the detection of a structural RNA that would otherwise not be detected. This is a replicate of the results from Gao et al. 2021.
The third example demonstrates a scenario where there is no significant covariation detected for HOTAIR, a long non-coding RNA using the unflanked mode. In the four examples, we run an unflanked mode search for a protein-coding query sequence to illustrate how R-scape can also identify protein-coding covariations.
1. xrRNA
Exoribonuclease-resistant RNA (xrRNA) are structural RNA elements that can resist degradation by exoribonucleases. These elements are often found in viral and coding and non-coding RNAs and play crucial roles in RNA stability and function, such as regulating gene expression or facilitating RNA transport.
In this example, we illustrate the usage of the following features of our method:
- Unflanking (“Single sequence”) mode
- How RNAhub deals with hits to the Rfam database
- How to interpret covariation results
Load the query sequence: For the xrRNA example, press the “load example” on the top menu under “xrRNA” to load the query sequence. In this example, we selected the query sequence corresponding to the intergenic region (IGR) of the Beet western yellows virus (BWYV) genomic sequence (GeneBank accession no. NC_004756) which was earlier reported to contain the xrRNA structure by Steckelberg et al. When this example is loaded, RNAhub will use the unflanked mode (“single sequence”) which just requires a query sequence. This should either be the sequence alone or can include a description line starting with “>”, conforming with the FASTA format.
Include job title and email address (optional): In addition to the sequence, the job title has been automatically listed as “xrRNA”. You can change the job title and also include an email address to which RNAhub will send the results once complete. This job only takes about 3-5 minutes to run because the query sequence is quite short and the viral database is quite small. For longer query sequences against eukaryotic genome databases, the entire RNAhub workflow can take several hours. Therefore, we recommend adding an email address so RNAhub can send you the results. If you do not include an email address, make sure to save/bookmark the tab that is running RNAhub.
Select genome database (required): You must select a database to search for homologs to the query sequence. In this case, the “Virus” database has already been selected.
Start search: Finally, click the “Search!” button to begin searching for homologs. Right after clicking search, the query sequence will be searched against known structural RNAs in the Rfam and PDB databases. Therefore, it will take a few seconds until the next screen is loaded.
Initial output:
The next page will show the following:
- Query sequence
- Hits to PDB database: In this case, there were no hits to PDB.
- Hits to Rfam database: In this case, there is a hit to Rfam. Therefore, the query sequence will be aligned to the top Rfam hit (lowest E-value). RNAhub will take the query sequence and the seed sequences from Rfam and align them together to create a structural alignment. R-scape is then run on this alignment to produce the structure below.
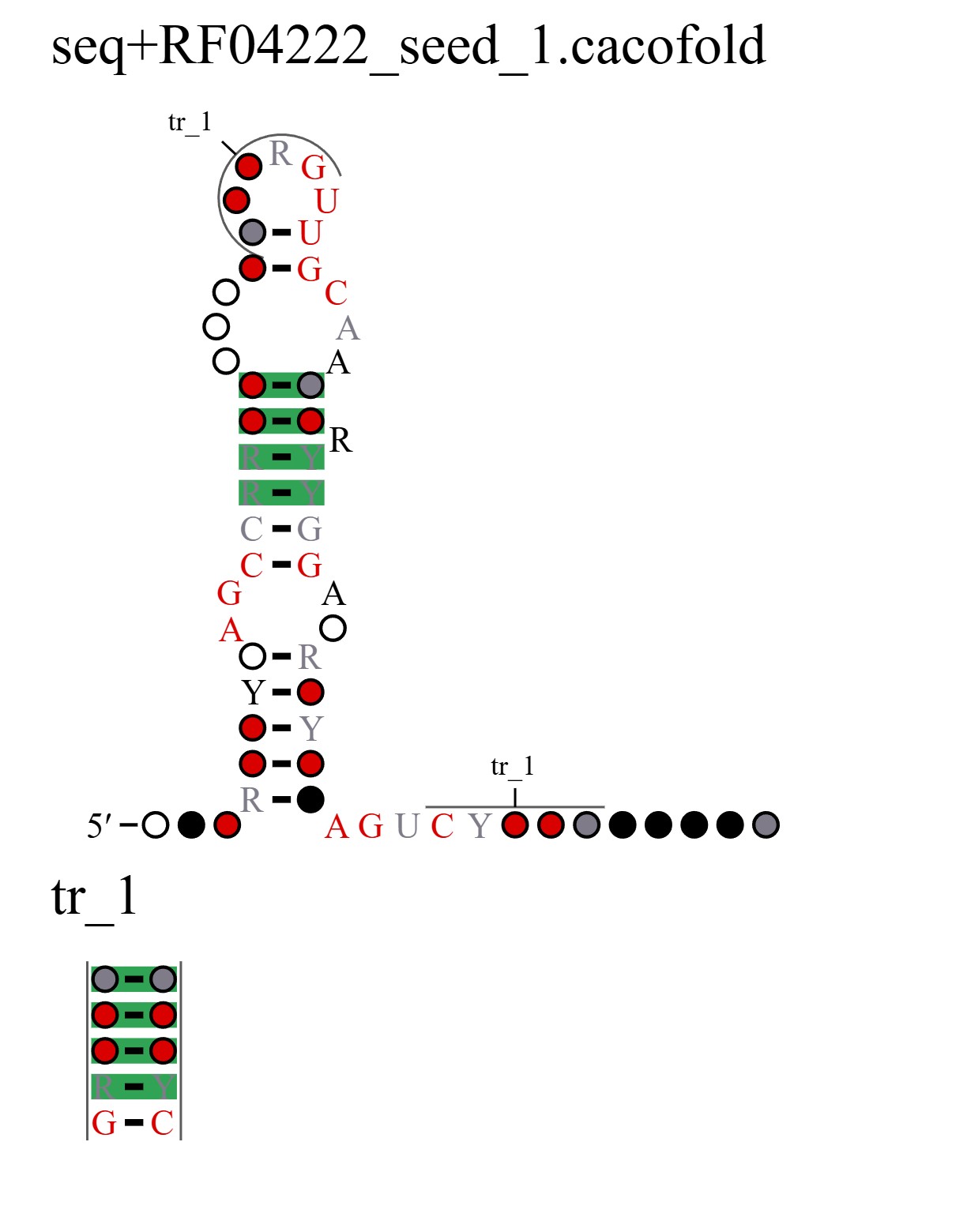
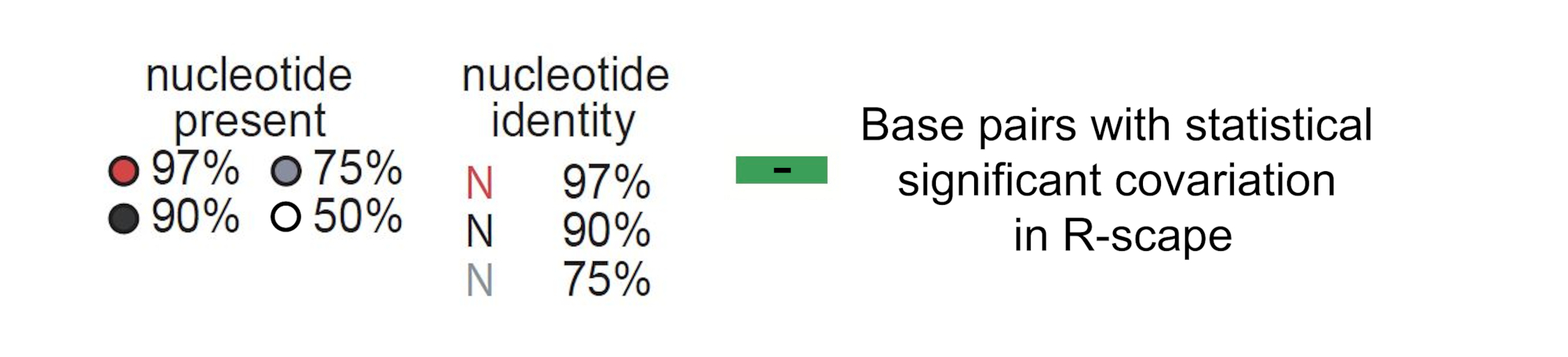
How to interpret the structure: The structure above is the predicted structure, with structural covariations highlighted in green. The structure is created using R2R (Weinberg et al. 2011). In addition to utilizing the sequence across homologs in the multiple sequence alignment, structural covariations are used to constrain the structure folding algorithm (see Rivas 2022 for more details on the CaCoFold algorithm). The primary sequence conservation of each nucleotide in the alignment is displayed according to the legend below the structure, with highly conserved residues displayed in red, black, gray, or white. A, C, G, and U refer to their respective nucleotides, Y standards for pyrimidine, and R for purine. Circles without letters refer to positions that are highly variable (without preference for particular nucleotides).
For example, if an A is found in 98% of aligned columns in the alignment, then it would be displayed as a red A. If A or G is found in 92% of aligned columns, then the color would be black and it would be displayed as R (for purine). If an aligned position is either U or C in 80% of the sequences, then it would be displayed as a gray Y (for pyrimidine). If an aligned column is highly variable and present in 60% of sequences, then it would be displayed as a white circle.
Interpreting the structure above, we observe 4 covariations in the main proposed stem-loop. Additionally, R-scape is able to identify pseudoknots. Separate from the main structure, there is a pseudoknot with 4 base pairs labeled as “tr_1”.
Covariation positions in the alignment:
Additionally, we provide the R-scape output in a tabular format, as shown below.
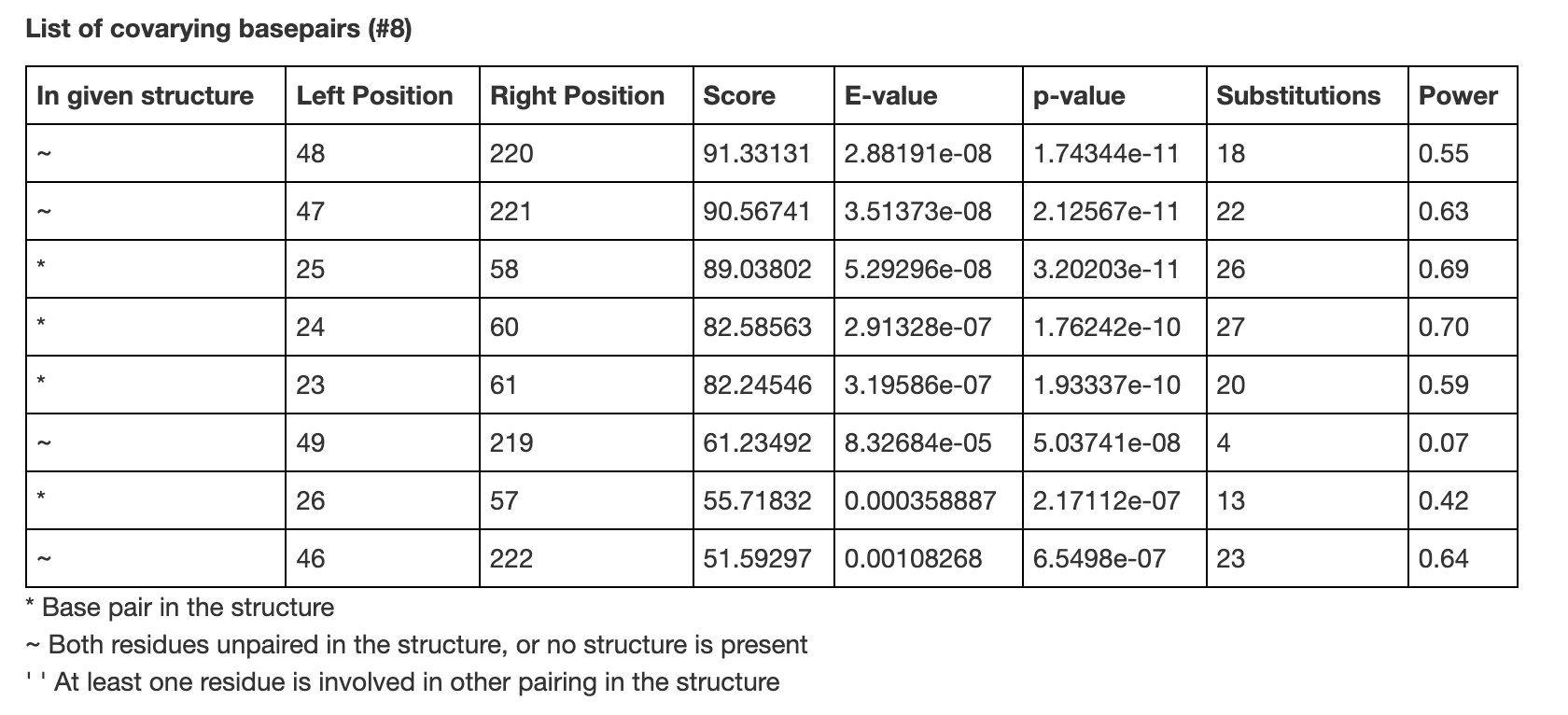
Notably, these covariations are displayed in terms of columns in the multiple sequence alignment. As only columns that are present in >50% of sequences are displayed in the R2R structure above, the columns that covary do not necessarily correspond to the residues in the R2R structure. Still, as the structure is displayed from the 5′ end to 3′ end, one can find which covarying base pairs correspond to each pair of columns.
Continuing the search:
At this point, since there is already a hit to Rfam, we could simply stop. Typically, if the user is interested in identifying whether a query sequence contains a structural RNA or not, then their answer would be clear at this point (i.e., there is a hit to the Rfam database). However, if the user wishes to continue the search despite a hit to Rfam, we can continue with the standard RNAhub workflow that performs three rounds of nhmmer and then uses that alignment to predict a secondary structure with R-scape.
We will click “continue the search” to continue through the entire unflanked mode in this tutorial and run the rest of the RNAhub workflow, which will take a few minutes.
Final output:
- R-scape on rfam alignment: This will be the output from the previous page, in which the query sequence was aligned with the detected Rfam hit.
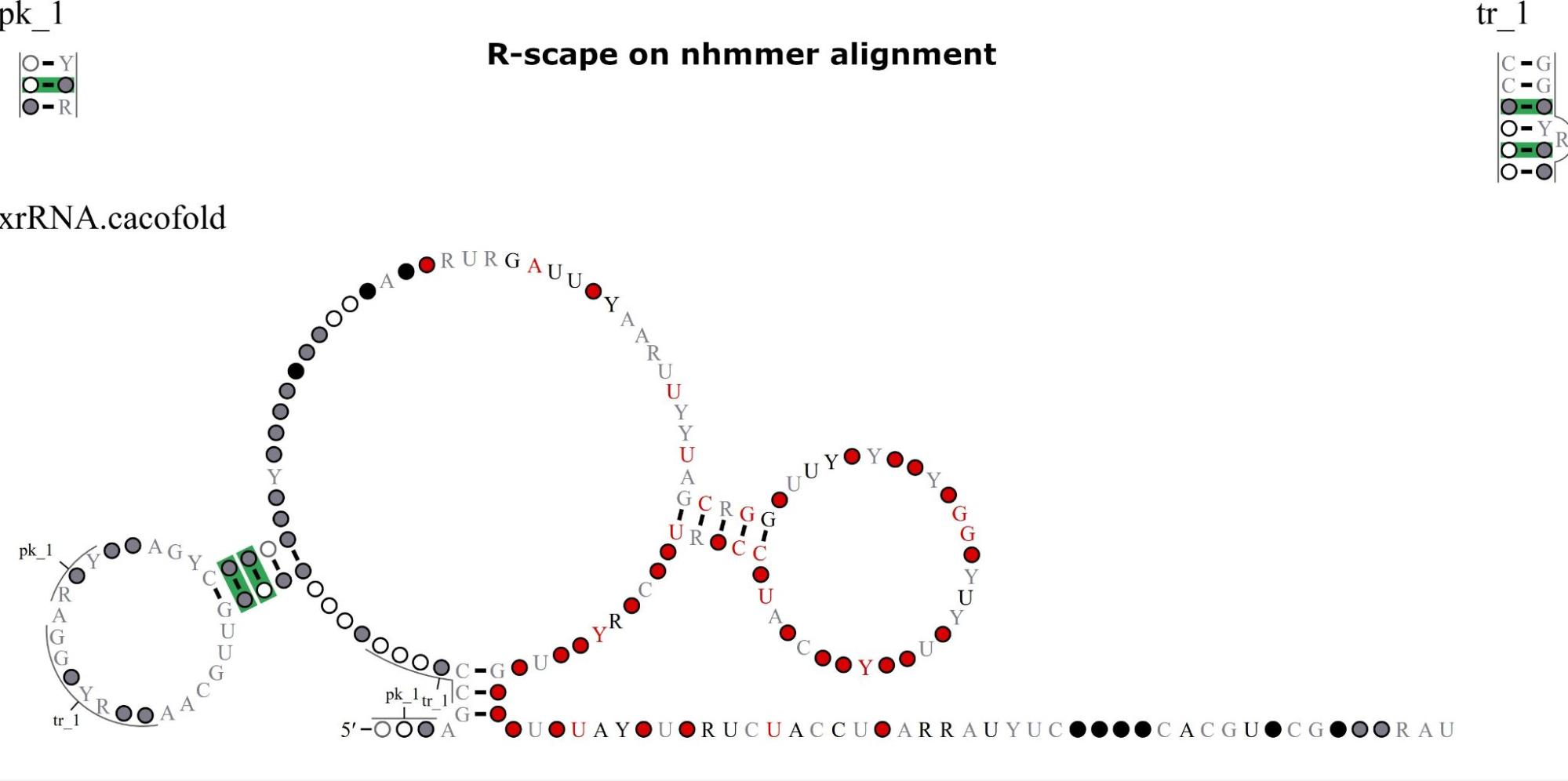
- R-scape on nhmmer alignment: This is the proposed structure based on the final nhmmer alignment that is passed to R-scape. Alignment statistics and the columns corresponding to covarying positions are also included.
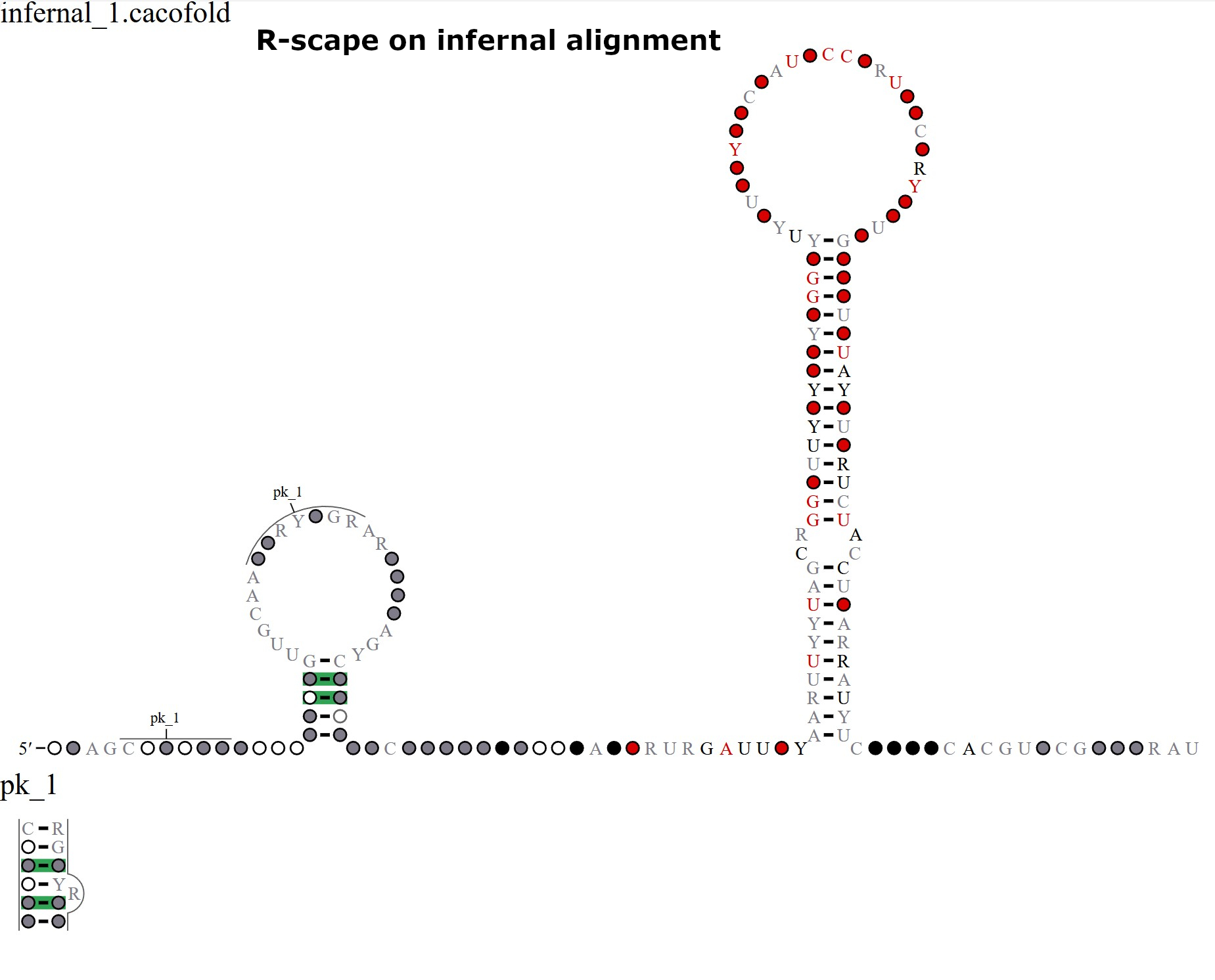
- R-scape on infernal alignment: Once the computation is complete, the next page will display the output. First, we will obtain an “R-scape on infernal alignment” option, in which the final iteration alignment was realigned using the R-scape/Cacofold predicted structure. This is only performed if there is at least one significant covariation detected at the nhmmer search level. The Infernal alignment can also be downloaded through the provided link. Alignment statistics are also provided, including the number of sequences and their similarity to each (percentage average identity). Additionally, the columns corresponding to covarying positions are also included.
Comparison of the different structures: While the three different structures have some similarities, there are also some notable differences. Across all alignments, we have a strong covariation signature suggesting that there is a strong likelihood that the query sequence contains a structural RNA. For some input alignments, we are able to detect pseudoknots. It is worth noting that while Infernal can improve structural covariation by realigning the sequences according to a predicted structure, it is not able to handle pseudoknots. Therefore, it is worth comparing the “R-scape on nhmmer alignment” and “R-scape on infernal alignment” (shown below) to see if there is a pseudoknot present in the nhmmer-based alignment that is lost when realigning with Infernal.
2. GLY1 3′ UTR using the flanking mode
In this example, we illustrate using the RNAhub flanking mode.
Why use the flanking mode?
RNAhub uses nhmmer, a primary sequence homology search tool, to build multiple sequence alignments. Structural RNAs, in contrast to protein-coding sequences, often diverge faster because their function lies in the maintenance of important base pairing interactions rather than codon similarity. The flanking mode can be used when a query sequence is associated with another adjacent region that may display higher primary sequence conservation. This flanking region facilitates the identification of homologs to query sequence in cases where the region pertaining to the query sequence itself diverges quickly but the flanking region evolves more slowly.
The best application of the flanking mode is typically when screening introns and untranslated regions of mRNAs for conserved structures. For example, many structural RNAs such as riboswitches are found in the 5’ UTR, intron, or 3’ UTR of mRNAs and perform cis-regulatory post-transcriptional functions.
GLY1 3′ UTR tutorial
We’ll walk through how to use the flanking mode on RNAhub using the example of the GLY1 3′ UTR discussed in Gao et al. 2021. This is a structure that is located in the 3’ UTR of the GLY1 gene in Saccharomyces cerevisiae.
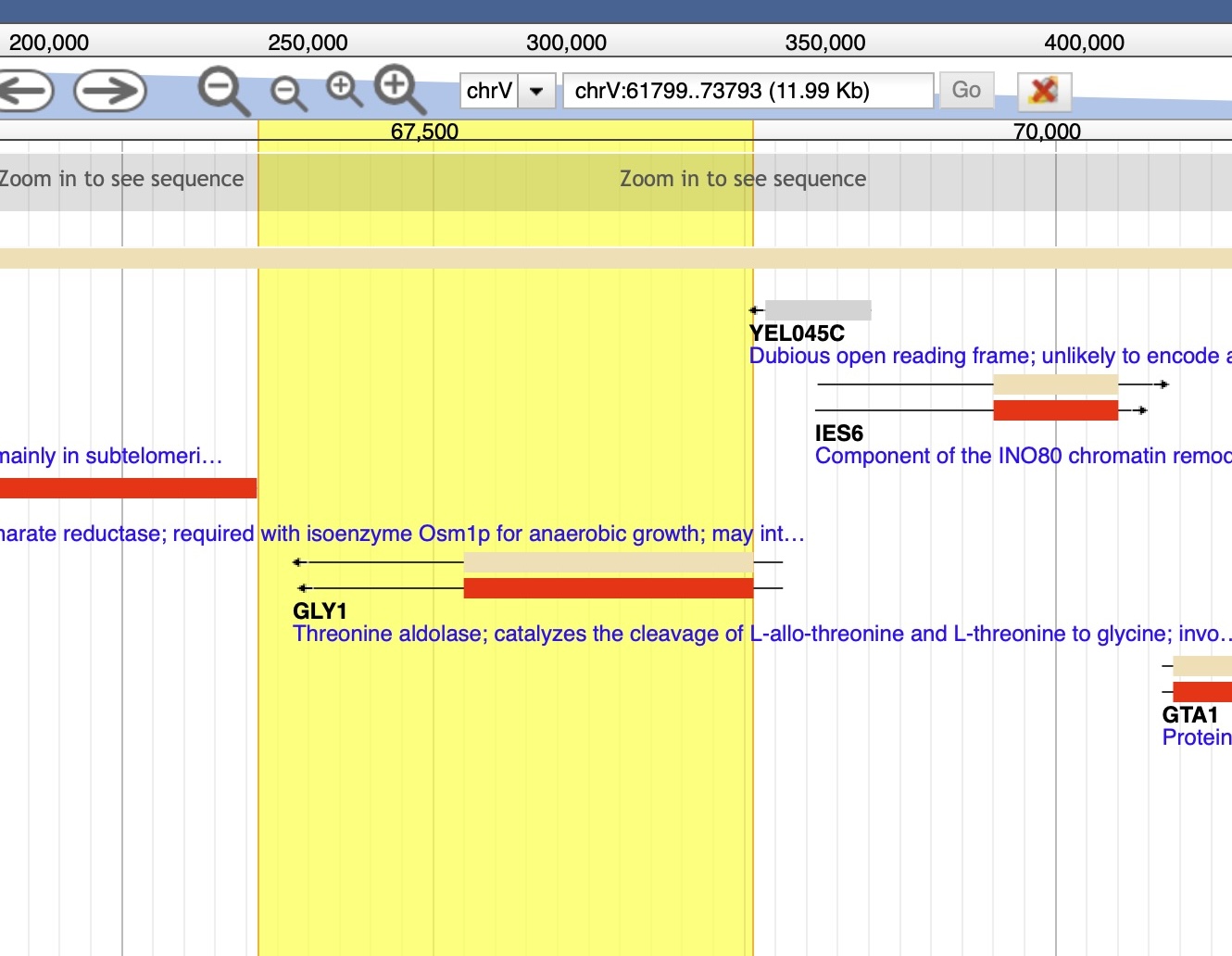
Figure. In yellow the whole sequence is marked and used for the search with a flanked sequence (search “0”, producing alignment “0.sto” in the project folder).
This genome browser visualization highlights a segment of chromosome V (chrV), spanning coordinates 61,799 to 73,793 (11.99 kb). The yellow-highlighted region includes the GLY1 gene and its untranslated region (UTR). GLY1 encodes threonine aldolase, an enzyme that catalyzes the cleavage of L-allo-threonine and L-threonine to glycine, playing a key role in amino acid metabolism. The annotation features directional arrows indicating transcription orientation and color-coded bars representing distinct genomic features.
There are two ways to use the flanking mode.
In the first approach (define start-end):
- Select “Flanking mode (define start-end)” from the three options. This is located right below the field for typing your email:
- Enter the entire sequence, which includes the query sequence AND the flanking sequence(s).
- Indicate the nucleotides corresponding to the query sequence, which is the region you are interested in building a final alignment of. The rest of the sequence is assumed to be the flanking regions and will be used for a single round of nhmmer at the beginning.
In the second approach (detect substring)
- Select “Flanking mode (detect substring)
- In the first text box, paste only the query sequence.
- In the second text box, paste the entire sequence (query sequence + flanking sequences). RNAhub will automatically compare these two sequences to determine which nucleotides correspond to the query sequence.
To get the input for either approach for the GLY1 example, simply press the button corresponding to them in the box at the top of the browser.

Once you have selected the input using either approach to specify the query sequence and flanking sequence, select the “Fungi” genome database for searching against and click “Search!”. This search will take a few hours to run. We recommend that you enter an email address, which will alert you when the run is finished.
Once the run is finished, you can compare your output to the intended output, which you can see by clicking the “Eye” button.
The output includes the following that can be downloaded:
- The raw output files and the computational commands performed at each step of the method
- The alignments, in Stockholm (.sto) format, at each nhmmer iteration
- The R-scape proposed structure, with significant covariations, highlighted
- A list of covarying base pairs. The covarying base pairs correspond to columns in the multiple sequence alignment.
- Alignment statistics for the final alignment that is passed into R-scape for covariation analysis.
- Any hits to Rfam or PDB databases, which include known structural RNAs.
- The entire input sequence, including flanking regions.
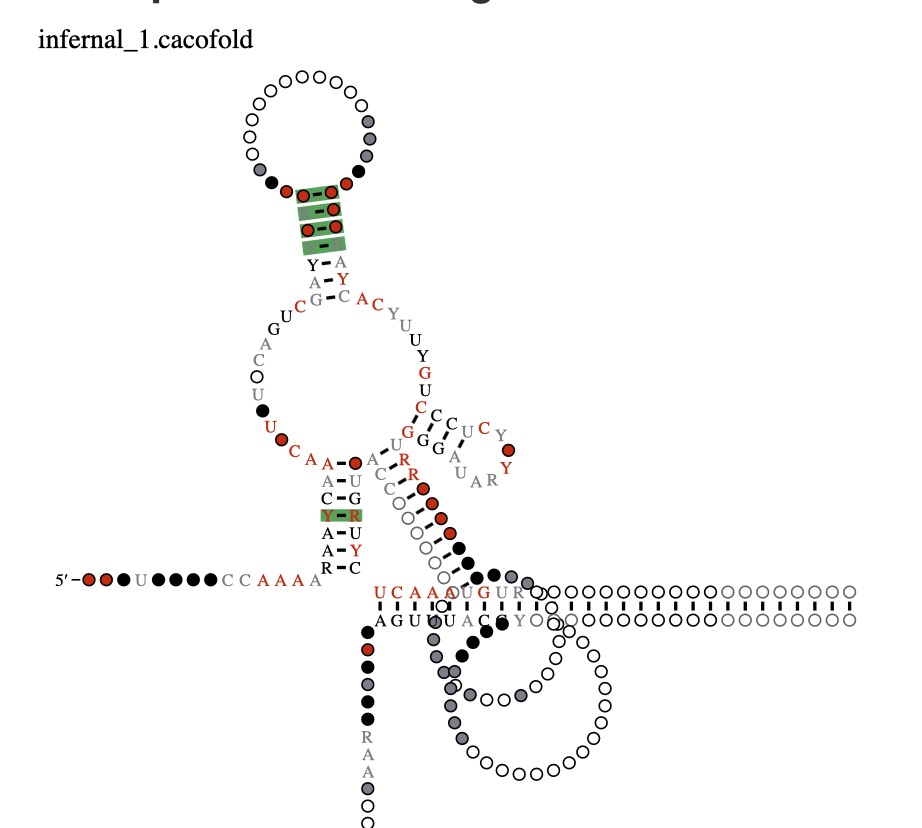
3. HOTAIR domain 1: long non-coding RNA without conserved structure
Here, we illustrate an example in which the HOTAIR domain 1 (D1) sequence from the human genome is queried against a database of 94 mammalian genomes using the unflanked mode (“Single sequence”). Clicking “load the example” under HOTAIR in the top menu will enter the query sequence into the RNAhub browser. Click “Search!” to perform the entire RNAhub workflow.
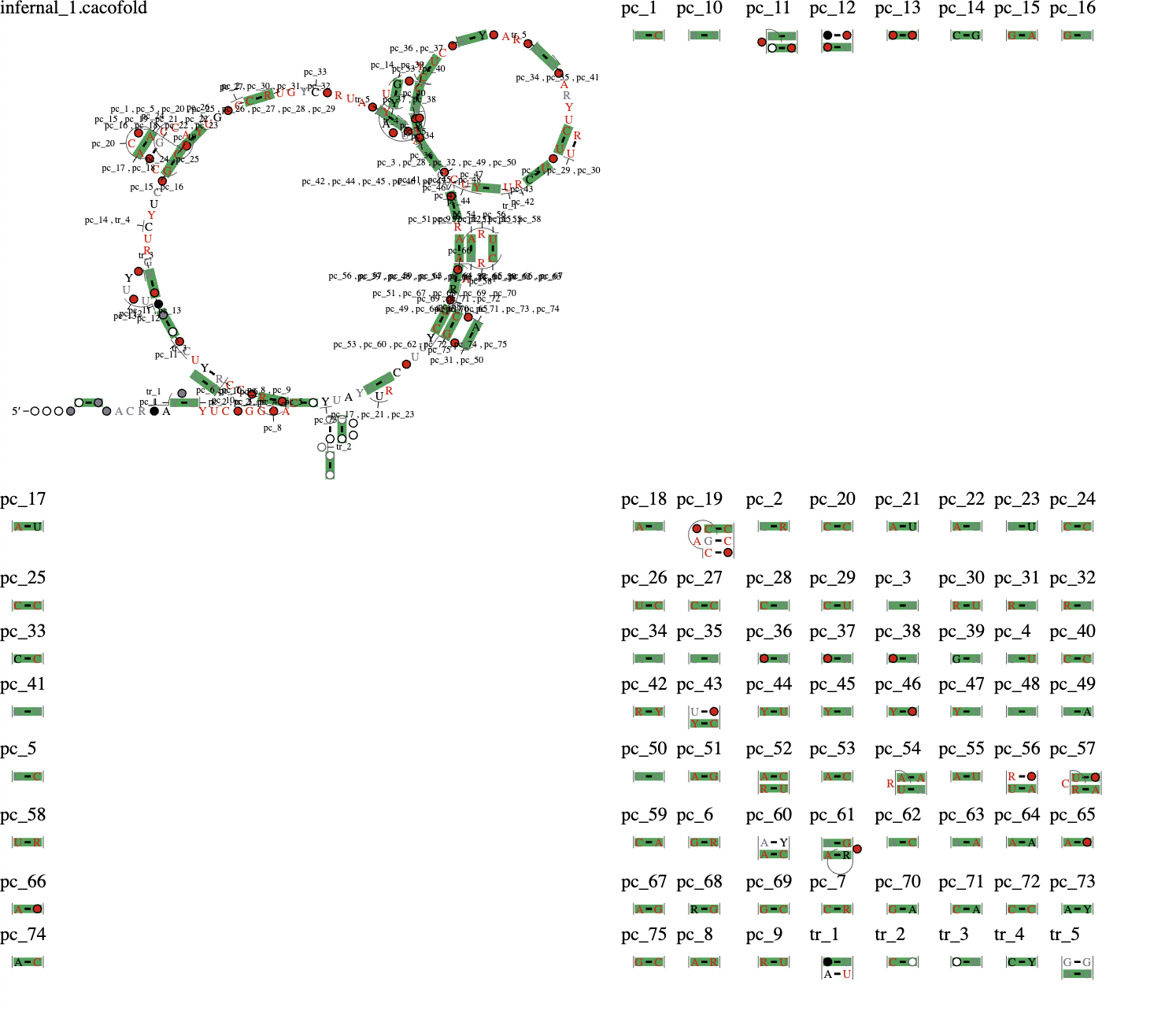
R-scape identifies base pairs that significantly covary beyond a phylogenetic null model. In addition to structural RNA covariation, another evolutionary signature beyond the phylogenetic null model is protein-coding covariations. These covariations reflect the codon triplet sequence and the evolution of protein-coding sequences in which nucleotides evolve across species in accordance with the maintenance of amino acid identity or functional similarity (see BLOSUM matrix). In contrast to structural RNA covariations, the strongest codon-related covariations are typically within-codon (<3 nucleotides apart). There are within-codon covariations that are >4 nucleotides, similar to structural RNA covariations but differ from structural RNA covariations because they do not conform with canonical base pairing (stem-loops, etc.). See Gao et al. 2022 for more information on our characterization of protein-coding covariations. In short, R-scape can also identify if a query sequence likely contains a region with protein-coding signatures. In this example, we query the human sequence of CWC15, a protein associated with the spliceosome. Clicking “load the example” under CWC15 in the top menu will enter the query sequence into the RNAhub browser. This will load YDR163W, the Saccharomyces cerevisiae sequence of CWC15. Then, click “Search!” to perform the entire unflanked RNAhub workflow to search the query sequence against the Fungi database. Due to the length of the query sequence, this search will take several hours. If you wish to see what the output is, click the “eye” under CWC15 in the top menu. In this output, you will see that no hits were detected against Rfam and PDB as expected since this query is not a structural RNA. The R-scape on the nhmmer alignment identifies numerous within-coding covariations, which are denoted as “pc_” in the structure. Below this structure is a tabular output of the columns that covary. Therefore, this sequence is most consistent with a covariation
signal
from a protein-coding region, rather than a structural RNA.
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. (2000). The Protein Data Bank. Nucleic Acids Res.
28
, 235–242. https://doi.org/10.1093/nar/28.1.235.
Camargo AP, Nayfach S, Chen I-MA, Palaniappan K, Ratner A, Chu K, et al. IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 2022;51:D733–43.
Gao, W., Yang, A., and Rivas, E. (2022). Thirteen dubious ways to detect conserved structural RNAs. Iubmb Life. https://doi.org/10.1002/iub.2694.
Gao, W., Jones, T.A., and Rivas, E. (2021). Discovery of 17 conserved structural RNAs in fungi. Nucleic Acids Res 49, gkab355-. https://doi.org/10.1093/nar/gkab355.
Ontiveros-Palacios, N., Cooke, E., Nawrocki, E.P., Triebel, S., Marz, M., Rivas, E., Griffiths-Jones, S., Petrov, A.I., Bateman, A., and Sweeney, B. (2024). Rfam 15: RNA families database in 2025. Nucleic Acids Res., gkae1023. https://doi.org/10.1093/nar/gkae1023.
Rivas, E., Clements, J., and Eddy, S.R. (2017). A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods
14
, 45–48. https://doi.org/10.1038/nmeth.4066.
Rivas, E. (2020). RNA structure prediction using positive and negative evolutionary information. Plos Comput Biol
16
, e1008387.
Steckelberg A-L, Vicens Q, Kieft JS. Exoribonuclease-Resistant RNAs Exist within both Coding and Noncoding Subgenomic RNAs. mBio. 2018;9:e02461-18.
Weinberg, Z. and Breaker, R.R., 2011. R2R-software to speed the depiction of aesthetic consensus RNA secondary structures.
BMC bioinformatics
,
12
, pp.1-9.
Wheeler, T.J., and Eddy, S.R. (2013).
nhmmer
: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. https://doi.org/10.1093/bioinformatics/btt403.
Due to the large size of the mammalian genome database and the length of the query sequence, this search will take several hours. If you wish to see what the output is, click the “eye” under HOTAIR in the top menu.
The final alignment created by the RNAhub has sequences from 92 genomes with an average percentage identity of 66%. The R-scape analysis of this alignment indicates that while the alignment expects 61 base pairs to covary, only 7 are found, and their arrangement does not support any RNA helix. Thus, the RNAhub alignment for HOTAIR-D1, as well as others before (Rivas, et. al. 2017), presents evidence against HOTAIR displaying a conserved RNA structure.
4. CWC15: example of protein-coding mRNA sequence
References